Strain and external fields
In this tutorial, we introduce the common procedure of applying strain and external fields on the
model. It is difficult to design common out-of-the-box user APIs that offer such functionalities
since they are strongly case-dependent. Generally, the user should implement these perturbations by
modifying model attributes such as orbital positions, on-site energies and hopping integrals. For
the primitive cell, it is straightforward to achieve this goal with the set_orbital and
add_hopping methods. The Sample class, on the contrary, does not offer such methods.
Instead, the user should work with the attributes directly. In the Sample class, orbital
positions and on-site energies are stored in the orb_pos and orb_eng attributes. Hopping
terms are represented with 2 attributes: hop_ind for orbital indices, and hop_eng
for hopping integrals. There is also an auxiliary attribute dr which holds the hopping vectors.
All the attributes are NumPy arrays. The on-site energies and hopping terms can be modified
directly, while the orbital positions should be changed via a modifier function. The hopping
vectors are updated from the orbital positions and hopping terms automatically, thus no need of
explicit modification.
As the example, we will investigate the propagation of wave function in a graphene sample. The
script can be found at examples/advanced/strain_fields.py. We begin with defining the
functions for adding strain and external fields, then calculate and plot the time-dependent wave
function to check their effects on the propagation. The essential packages of this tutorial can be
imported with:
import math
from typing import Callable, Tuple
import numpy as np
from numpy.linalg import norm
import tbplas as tb
Functions for strain
Strain will introduce deformation into the model, changing both orbital positions and hopping integrals. It is a rule that orbital positions should not be modified directly, but through a modifier function. We consider a Gaussian bump deformation, and define the following functions to generate the modifier:
1def gaussian(orb_pos: np.ndarray,
2 center: np.ndarray,
3 sigma: Tuple[float, float] = (0.5, 0.5),
4 extent: Tuple[float, float] = (1.0, 1.0)) -> Dict[str, np.ndarray]:
5 """
6 Generate normalized Gaussian distribution based on the orbital positions.
7
8 :param orb_pos: (num_orb, 3) float64 array, orbital positions in nm
9 :param center: Cartesian coordinate of the Gaussian center in nm
10 :param sigma: width of Gaussian along x and y directions in nm
11 :param extent: factors controlling Gaussian extent along x and y directions
12 :return: the Gaussian distribution
13 """
14 dx = (orb_pos[:, 0] - center[0]) * extent[0]
15 dy = (orb_pos[:, 1] - center[1]) * extent[1]
16 amp = np.exp(- dx ** 2 / (2 * sigma[0] ** 2) - dy ** 2 / (2 * sigma[1] ** 2))
17 amp /= (2 * math.pi * np.prod(sigma))
18 result = {"dx": dx, "dy": dy, "amp": amp}
19 return result
20
21
22def make_deform(scale: Tuple[float, float] = (0.5, 0.5), **kwargs) -> Callable:
23 """
24 Generate Gaussian-shaped deformation function as orb_pos_modifier.
25
26 :param scale: scaling factors for deformation along xOy and z directions
27 :param kwargs: arguments for 'gaussian' function
28 :return: deformation function
29 """
30 def _deform(orb_pos):
31 g = gaussian(orb_pos, **kwargs)
32 orb_pos[:, 0] += g["dx"] * g["amp"] * scale[0]
33 orb_pos[:, 1] += g["dy"] * g["amp"] * scale[0]
34 orb_pos[:, 2] += g["amp"] * scale[1]
35 return _deform
The arguments center, sigma and extent in function gaussian control the location, width and
extent of the Gaussian distribution, respectively. For example, if extent is set to \((1.0, 0.0)\),
the distribution will become one-dimensional which varies along \(x\)-direction while remains constant
along \(y\)-direction. The scale argument in function make_deform specifies the scaling factors
for in-plane and out-of-plane displacements. The make_deform function returns another function as the
modifier, which updates the orbital positions in place according to the following expression:
where \(\mathbf{r}_i\) is the position of \(i\)-th orbital, \(\Delta_i\) is the displacement, \(s\) is the scaling factor, \(\parallel\) and \(\perp\) are the in-plane and out-of-plane components. The location, width and extent of the Gaussian bump are denoted as \(\mathbf{c}_0\), \(\sigma\) and \(\eta\), respectively.
In addition to the orbital position modifier, we also need to update hopping integrals
1def update_hop(sample: tb.Sample) -> None:
2 """
3 Update hopping terms in presence of deformation.
4
5 :param sample: Sample to modify
6 :return: None.
7 """
8 sample.init_hop()
9 for i, rij in enumerate(sample.dr):
10 sample.hop_eng[i] = calc_hop(rij)
As we will make use of the hopping terms and vectors, we should call the init_hop method to initialize the
attributes. Similar rule holds for the on-site energies and orbital positions. Then we loop over the hopping
terms to update the integrals in hop_eng according to the vectors in dr with the calc_hop function,
which is defined as:
1def calc_hop(rij: np.ndarray) -> float:
2 """
3 Calculate hopping parameter according to Slater-Koster relation.
4 See ref. [2] for the formulae.
5
6 :param rij: (3,) array, displacement vector between two orbitals in NM
7 :return: hopping parameter in eV
8 """
9 a0 = 0.1418
10 a1 = 0.3349
11 r_c = 0.6140
12 l_c = 0.0265
13 gamma0 = 2.7
14 gamma1 = 0.48
15 decay = 22.18
16 q_pi = decay * a0
17 q_sigma = decay * a1
18 dr = norm(rij).item()
19 n = rij.item(2) / dr
20 v_pp_pi = - gamma0 * math.exp(q_pi * (1 - dr / a0))
21 v_pp_sigma = gamma1 * math.exp(q_sigma * (1 - dr / a1))
22 fc = 1 / (1 + math.exp((dr - r_c) / l_c))
23 hop = (n**2 * v_pp_sigma + (1 - n**2) * v_pp_pi) * fc
24 return hop
Functions for external fields
The effects of external electric field can be modeled by adding position-dependent potential to the on-site energies. We consider a Gaussian-type scattering potential described by
and define the following function to add the potential to the sample
1def add_efield(sample: tb.Sample, v_pot: float = 1.0, **kwargs) -> None:
2 """
3 Add Gaussian-shaped electric field to the sample.
4
5 :param sample: sample to add the field
6 :param v_pot: electric field intensity in eV
7 :param kwargs: arguments for 'gaussian'
8 :return: None.
9 """
10 sample.init_orb_pos()
11 sample.init_orb_eng()
12 sample.orb_eng += v_pot * gaussian(sample.orb_pos, **kwargs)["amp"]
The argument v_pot specifies \(V_0\). Similar to update_hop, we need to call
init_orb_pos and init_orb_eng to initialize orbital positions and on-site energies before
accessing them. Then the position-dependent scattering potential is added to the on-site energies.
The effects of magnetic field can be modeled with Peierls substitution. For homogeneous magnetic
field perpendicular to the \(xOy\)-plane along \(-z\) direction, the Sample
class offers an API apply_magnetic_field, which follows the Landau gauge of vector potential
\(\mathbf{A} = (By, 0, 0)\) and updates the hopping terms as
where \(B\) is the intensity of magnetic field, \(\mathbf{r}_i\) and \(\mathbf{r}_j\) are the positions of \(i\)-th and \(j\)-th orbitals, respectively.
Initial wave functions
The initial wave function we consider here as an example for the propagation is a Gaussian wave-packet, which is defined by
1def init_wfc_gaussian(sample: tb.Sample, **kwargs) -> np.ndarray:
2 """
3 Generate Gaussian wave packet as initial wave function.
4
5 :param sample: sample for which the wave function shall be generated
6 :param kwargs: arguments for 'gaussian'
7 :return: initial wave function
8 """
9 sample.init_orb_pos()
10 wfc = np.array(gaussian(sample.orb_pos, **kwargs)["amp"], dtype=np.complex128)
11 wfc /= np.linalg.norm(wfc)
12 return wfc
Note that the wave function should be a complex vector whose length must be equal to the number of orbitals. Also, it should be normalized before being returned.
Propagation of wave function
We consider a rectangular graphene sample with \(50\times20\times1\) primitive cells, as shown in Fig. 1(a). We begin with defining some geometric parameters:
1prim_cell = tb.make_graphene_rect()
2dim = (50, 20, 1)
3pbc = (True, True, False)
4x_max = prim_cell.lat_vec[0, 0] * dim[0]
5y_max = prim_cell.lat_vec[1, 1] * dim[1]
6wfc_center = (x_max * 0.5, y_max * 0.5)
7deform_center = (x_max * 0.75, y_max * 0.5)
Here dim and pbc define the dimension and boundary condition. x_max and y_max are
the lengths of the sample along \(x\) and \(y\) directions. The initial wave function will
be a Gaussian wave-packet located at the center of the sample given by wfc_center.
The deformation and scattering potential will be located at the center of right half of the sample,
as specified by deform_center and shown in Fig. 1(b)-(c).

Top and side views of (a) pristine graphene sample and (b) sample with deformation. (c) Plot of on-site energies of graphene sample with scattering potential.
We firstly investigate the propagation of a one-dimensional Gaussian wave-packet in pristine sample, which is given by
1# Prepare the sample and inital wave function
2sample = tb.Sample(tb.SuperCell(prim_cell, dim, pbc))
3psi0 = init_wfc_gaussian(sample, center=wfc_center, extent=(1.0, 0.0))
4
5# Propagate the wave function
6time_log = np.array([0, 16, 32, 64, 128])
7solver = tb.TBPMSolver(sample)
8solver.config.rescale = 10.0
9solver.config.num_time_steps = 128
10solver.config.wft_wf0 = psi0
11solver.config.wft_time_save = time_log
12psi_t = solver.calc_wft()
13
14# Visualize the time-dependent wave function
15vis = tb.Visualizer()
16for i in range(len(time_log)):
17 vis.plot_wfc(sample, np.abs(psi_t[i])**2, cmap="hot", scatter=False)
As the propagation is performed with the calc_wft function of TBPMSolver class, it follows
the common procedure of TBPM calculation. We need to set the rescaling factor (rescale), the
number of steps of propagation (num_time_steps). The initial state is specified by wft_wf0.
We propagate the wave function by 128 steps, and save the snapshots in psi_t at the time steps
specified by wft_time_save. The snapshots are then visualized by the plot_wfc function of
Visualizer class, as shown in Fig. 2(a)-(e), where the wave-packet diffuses freely, hits the
boundary and forms interference pattern.
We then add the bump deformation to the sample, by assigning the modifier function to the supercell
and calling update_hop to update the hopping terms
deform = make_deform(center=deform_center)
sample = tb.Sample(tb.SuperCell(prim_cell, dim, pbc, orb_pos_modifier=deform))
update_hop(sample)
The propagation of wave-packet in deformed graphene sample is shown in Fig. 2(f)-(j). Obviously, the wave function gets scattered by the bump. Although similar interference pattern is formed, the propagation in the right part of the sample is significantly hindered, due to the increased inter-atomic distances and reduced hopping integrals at the bump.
Similar phenomena are observed when the scattering potential is added to the sample by
add_efield(sample, center=deform_center)
The time-dependent wave function is shown in Fig. 2(k)-(o). Due to the higher on-site energies, the probability of emergence of electron is suppressed near the scattering center.
As for the effects of magnetic field, it is well known that Landau levels will emerge in the DOS. The analytical solution to Schrodinger’s equation for free electron in homogeneous magnetic field with \(\mathbf{A}=(By, 0, 0)\) shows that the wave function will propagate freely along \(x\) and \(z\)-directions while oscillates along \(y\)-direction. To simulate this process, we apply the magnetic field to the sample by
sample.apply_magnetic_field(50)
The snapshots of time-dependent wave function are shown in Fig. 2(p)-(t). The interference pattern is similar to the case without magnetic field, as the wave function propagates freely along \(x\) direction. However, due to the oscillation along \(y\)-direction, the interference pattern gets distorted during the propagation. These phenomena agree well with the analytical results.
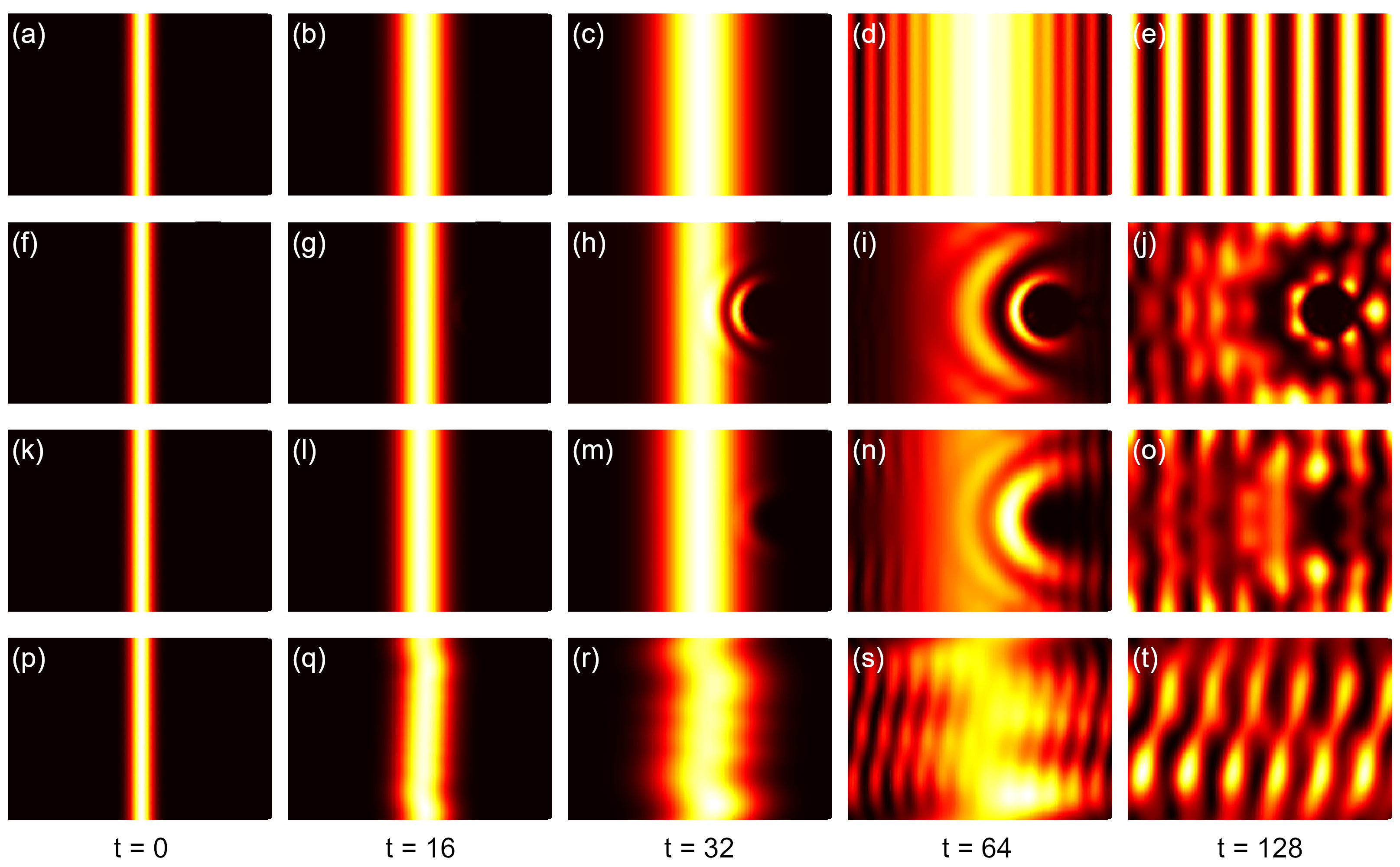
(a)-(e) Propagation of one-dimensional Gaussian wave-packet in pristine graphene sample. (f)-(j) Propagation in graphene sample with deformation, (k)-(o) with scattering potential and (p)-(t) with magnetic field of 50 Tesla.